CF (Cystic Fibrosis)
Cystic fibrosis (CF) is one of the most common serious genetic conditions that affects mostly the lungs and digestive system.
What is cystic fibrosis?
What are the causes of cystic fibrosis?
What are the signs and symptoms of cystic fibrosis?
What are the possible tests to detect cystic fibrosis?
What are the possible procedures and treatments for cystic fibrosis?
What is the future plan if you have cystic fibrosis?
What is cystic fibrosis?
Cystic fibrosis (CF) is one of the most common serious genetic conditions which affects about 1 in 1600 people at birth. CF can affect almost every body system and is associated with a reduced lifespan, though this is improving with recent advances in medical care.
What are the causes of cystic fibrosis?
CF is a genetic condition, which means you are born with it. In order for CF to develop, both parents must be carriers of the gene that produces CF. When both parents carry the CF gene, there is a 25% chance that their baby will have CF.
Both boys and girls have an equal chance of developing CF, however it is more common in Caucasians than other ethnicities.
What are the signs and symptoms of cystic fibrosis?
CF mainly affects the lungs and the digestive system. People with CF produce excessive mucus that is ‘glue-like’ in nature. Sweat, tears and saliva, as well as digestive juices, can also be thicker and stickier than normal.
In people with CF, mucus builds up in the lungs and blocks the airways, making it harder to breathe. The excess mucus also means bacteria can grow easily, increasing the risk and frequency of lung infections.
The image below illustrates cystic fibrosis in the lungs. (Click image to enlarge)
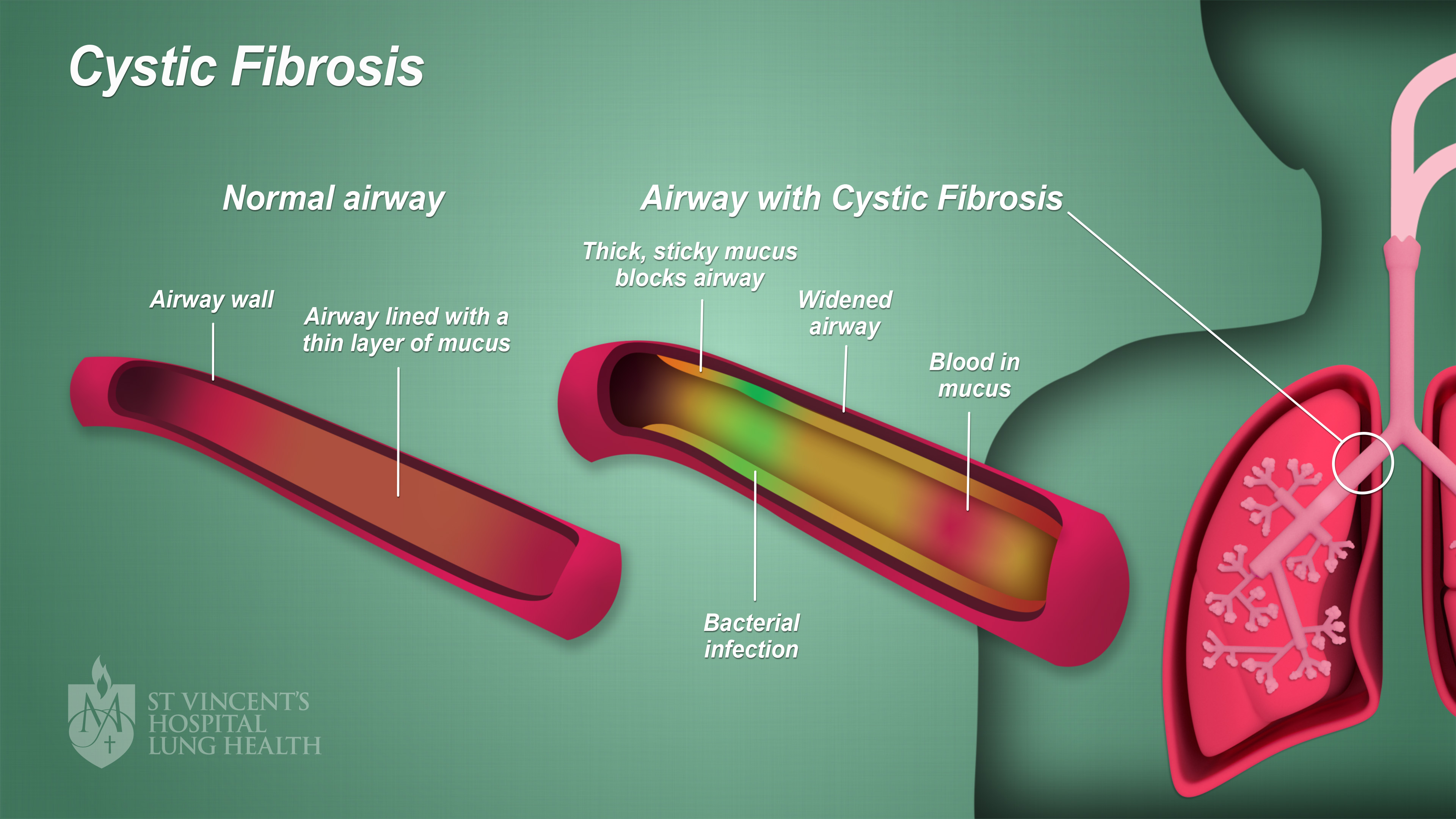
Excessive mucus also damages the digestive system, particularly the pancreas, by stopping special enzymes (which break down your food) from reaching your gut. The pancreas also may not produce enough insulin so that diabetes is common. Symptoms of CF can begin at birth, though symptoms may appear weeks, months or even years after birth, too.
The most common symptoms of CF include:
- Bowel movements that are smelly or greasy
- Constipation (unable to pass a bowel movement)
- Continual coughing or wheezing with difficulty breathing that worsens over time
- Failure to put weight on despite eating an adequate diet
- Pneumonia (lung infection) or sinusitis (sinus infection)
- Skin that is salty to taste.
As time progresses, additional symptoms of cystic fibrosis can include:
- A chronic productive cough
- Bronchiectasis, a condition where the airways are enlarged due to wall damage
- Chronic congestion of the nasal passages resulting in ongoing sinus infections
- Diabetes
- Disease of the liver
- Gallstones
- Ongoing infections of the lung.
What are the possible tests to detect cystic fibrosis?
There are a number of different testing methods available to screen and diagnose cystic fibrosis. These tests typically involve testing either your sweat or your blood.
- Carrier testing – is where your DNA is analysed to see if you have the gene which causes CF; this can be done via a blood or saliva sample (if both you and your partner are carriers of the CFTR gene, there is a risk that your baby will be born with CF, and carrier testing is also available to women early in their pregnancies and their partners)
- Chloride sweat test – if a genetic or blood test suggests CF then a ‘sweat test’ will be carried out; this is the most reliable test for CF and involves collecting sweat on a pad and analysing the salt level of the sweat (high levels of salt confirm a diagnosis of CF)
- Newborn screening – also known as a ‘heel prick’ test, this test is a blood test done a couple of days after the baby is born; a few drops of the baby’s blood is tested to see if its’ pancreas is functioning correctly.
What are the possible procedures and treatments for cystic fibrosis?
While there is currently no cure for CF, there are a number of treatment options which aim to prevent and manage infections of the lung as well as maintain function of the digestive system.
Treatment typically involves:
- Exercises – to loosen mucus in the lungs as well as build strength
- Insulin – for diabetes
- Meal plans – that have high levels of calories, fat and salts to help increase nutrition levels
- Medications – such as mucus thinners, laxatives, anti-inflammatories, antibiotics and bronchodilators (which assist in opening the airways)
- Regular, daily chest physiotherapy (CPT) – a special type of therapy involving a ‘pounding’ or vibration on the chest, which assists in loosening and drainage of mucus from the lungs
- Supplements – including; vitamins (in particular fat-soluble vitamins A, D, E and K), salts and digestive enzyme capsules (for digestion and absorption of food)
- Surgery – may be required as a result of complications due to cystic fibrosis and can include: sinus and bowel surgery, as well as lung transplants.
What is the future plan if you have cystic fibrosis?
Whilst CF is a serious, life-long condition, many treatments enable people who have CF to live much longer than in previous years.
New medications and combinations add great hope for certain patients with CF to target the specific abnormality.
Your CF specialist may discuss the potential role of lung transplant with you. A number of St Vincent’s patients have had very successful outcomes and are now living well more than 20 years after their lung transplant.
Following your individual treatment plan as well as regular visits to a Cystic Fibrosis Treatment Centre will ensure you have the best quality of life possible.